This page was produced as an assignment for Genetics 564, an undergraduate capstone course at The University of Wisconsin - Madison.
Introduction
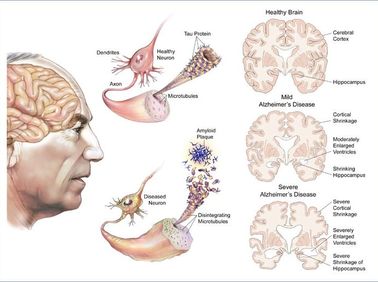
Alzheimer's is a neurodegenerative disease. As a form of dementia, Alzheimer's is a progressive disease that results in loss of memory, language, recognition, and motor skills. The two hallmarks of the disease that are found in the brain are Tau Tangles and Amyloid Beta Plaques. For the formation of amyloid plaques, the widely accepted Amyloid Cascade Hypothesis shapes our current understanding of the disease.
|
|
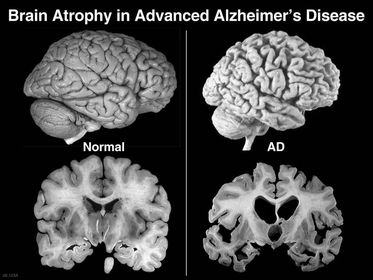
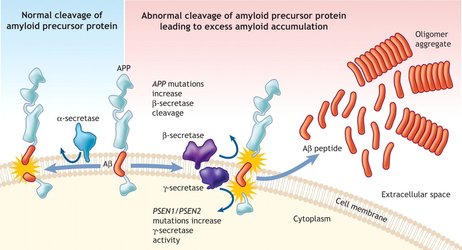
Amyloid precursor protein (APP) is an integral protein that is central to the Amyloid Cascade Hypothesis and the gene that codes for that protein is associated with Alzheimer's. In the cascade, APP begins as an integral membrane protein located in the synapses of the brain. Under normal conditions, APP is cleaved and the integrated Amyloid Beta portion of the protein is expelled into the extracellular environment. Normally Amyloid Beta is cleared by microglial cells, but in Alzheimer's Amyloid Beta accumulates in to large amyloid plaques that lead to brain atrophy. The cause of the change from healthy cleavage and clearance to the disease state is still unknown but many cases of Alzheimer's are associated with the increased regulation of APP.
|
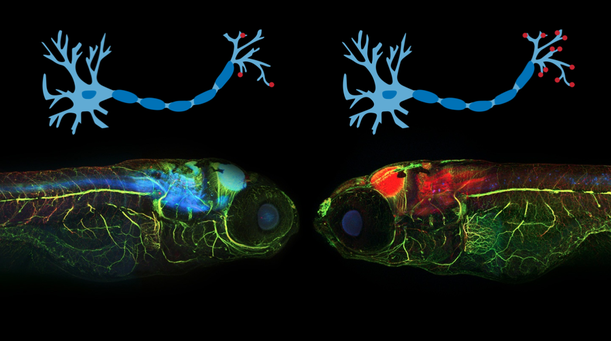
Designing a model of the amyloid cascade in zebrafish allows us to quantify the both the expression of APP and the accumulation of amyloid plaques by staining
|
|
In Alzheimer's, increase transcription levels of APP are associated with brain atrophy. Previous studies [1, 2, 3] indicate that the upstream promoter regions -3830, -1023, -534, -479, -369 play a significant role in regulating protein transcription. Clustal Omega base pair alignment indicates that those candidate base pairs are highly conserved across model organisms. This leads us to my primary gap in knowledge: if we mutagenize those candidate regions, will that increase the transcription levels of APP in the neuronal synapses?
Specific Aim 1
Aim 1 is to determine which conserved base pairs in the promoter region are necessary for APP transcription in neurons I will use CRISPR/CAS9 to mutagenize the promoter and observe each region’s affect on APP transcription. The change in transcription levels will be monitored quantitatively with gels and protein spectrometry and also quantitatively with a behavioral test. I hypothesize that the mutagenesis will induce increased transcription of APP as indicated in the gel and will result in memory loss for the zebrafish. I will use the following approach to determine which regions of the promoter region regulate APP transcription.
- Design a vector for zebrafish containing the APP promoter, coding regions, and a histidine tag.
- Use CRISPR/Cas9 in the 5’ promoter region at sites -369, -479, -534, -1023, and -3830.
- Quantify the amount of APP transcribed with protein spectrometry, gel electrophoresis, and a zebrafish behavioral maze test.
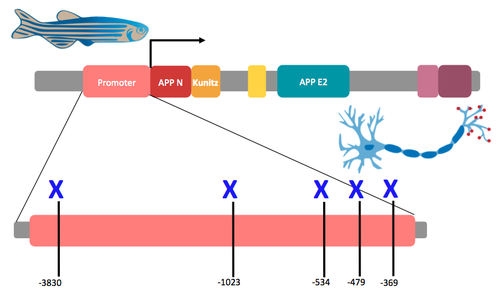
Overview of Aim 1.
Specific Aim 2
In the Amyloid Cascade that APP is cleaved from the membrane, releasing Amyloid Beta, which in AD accumulate into large amyloid plaques that cause damage to the brain. Does the increased promoter regulation from AIM 1 change the solubility of those plaques or does it also change the solubility of the resulting AB? Aim 2 is to determine if mutagenesis performed in Aim 1 affects the solubility of the resulting amyloid plaques. I hypothesize that mutagenesis will have no impact on plaque solubility because to promoter region. To test the solubility of the promoter mutant amyloid plaques with the Alzheimer’s drugs Florabetaben, Methionine Sulfoxide, and Flutemetamol. These drugs treat Alzheimer’s by solubilizing amyloid plaques in the brain. I will use the following approach to determine if the mutagenesis in the promoter affects the solubility of the resulting amyloid plaques.
- Use the zebrafish model created in Aim 1 to plate neurons
- Treat each group with one of the Alzheimer’s drugs
- Quantify the amount of remaining amyloid plaque with protein spectrometry, gel electrophoresis, and a zebrafish behavioral maze test.
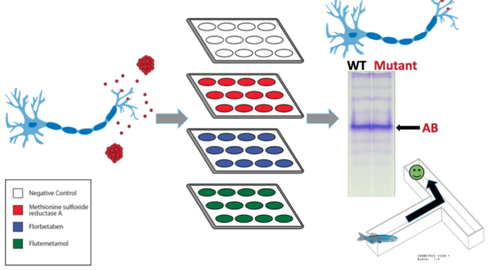
Specific Aim 3
When comparing the predicted phosphorylation sites of APP using NetPhos, I noticed this highly activated. This highly activated sequence contains the Beta region that is cleaved into the extracellular environment. This indicates that increased transcription of APP would proportionally increase the phosphorylation levels of the Kunitz domain. Aim 3 is to determine the phosphorylation levels of mutant APP promoter protein product in the brain tissue. I hypothesize that there will be a increase in phosphorylation levels in the mutant APP promoter protein product that is proportional to APP expression because of the higher abundance of APP cleavage sites. I will use SILAC-based mass spectrometry to quantify the phosphorylation present in mutant APP promoter protein products. Mutant proteins will be compared relative to the WT promoter protein product. I will use the following approach to determine the phosphorylation levels of the mutant promoter protein products.
- Feed the zebrafish model created in Aim 1 with food containing a heavy isotope of Carbon.
- Quantify the phosphorylation levels with mass spectrometry.
- Compare the phosphorylation levels to the level of APP transcription
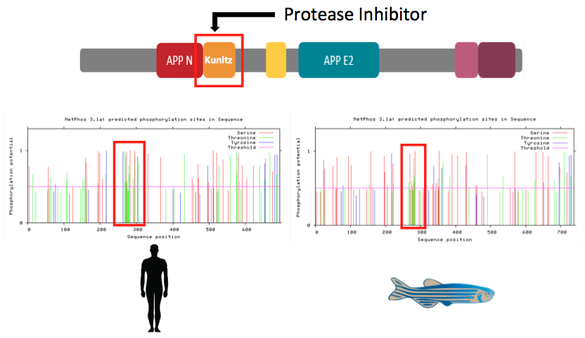
Conclusions and Future Directions
Characterizing the upstream promoter region for APP will improve our understanding of how APP regulation leads to the accumulation of amyloid plaques in Alzheimer’s disease. Knowing which sites in the promoter are responsible for this regulation will lead to new ways to prevent the accumulation of Aβ in patients with Alzheimer’s and patients who are at risk for the disease.
References
[1] Guyant-Maréchal, L. et al. 2007. Variations in the APP gene promoter region and risk of Alzheimer disease. Neurology. 68: 684-687.
[2] Theuns, J. et al. 2006. Promoter Mutations that Increase Amyloid Precursor-Protein Expression are Associated with Alzheimer Disease. The American Journal of Human Genetics. 78: 936-946.
[3] Bailey, J. et al. 2011. Functional activity in of the novel Alzheimer’s amyloid β-peptide interacting domain (AβID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes in amyloidogenesis. Gene. 488: 1-2: 13-22.
[2] Theuns, J. et al. 2006. Promoter Mutations that Increase Amyloid Precursor-Protein Expression are Associated with Alzheimer Disease. The American Journal of Human Genetics. 78: 936-946.
[3] Bailey, J. et al. 2011. Functional activity in of the novel Alzheimer’s amyloid β-peptide interacting domain (AβID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes in amyloidogenesis. Gene. 488: 1-2: 13-22.
Final Draft and Presentations
| eneale_presentation_draft_1.pptx |
| eneale_part1part2_4-4-17.pptx |
| eneale_final_presentation.pdf |
